Escherichia coli : organisme modèle de laboratoire
A la recherche de leurs ancêtres
Fun fact : Les bactéries sont apparues sur terre, il y a 3,8 milliards d’années, soit 3,5 milliards d’années avant les humains ! Beaucoup ont disparu mais l’ADN des cellules actuelles conserve une trace de la façon dont ces organismes ont évolué.
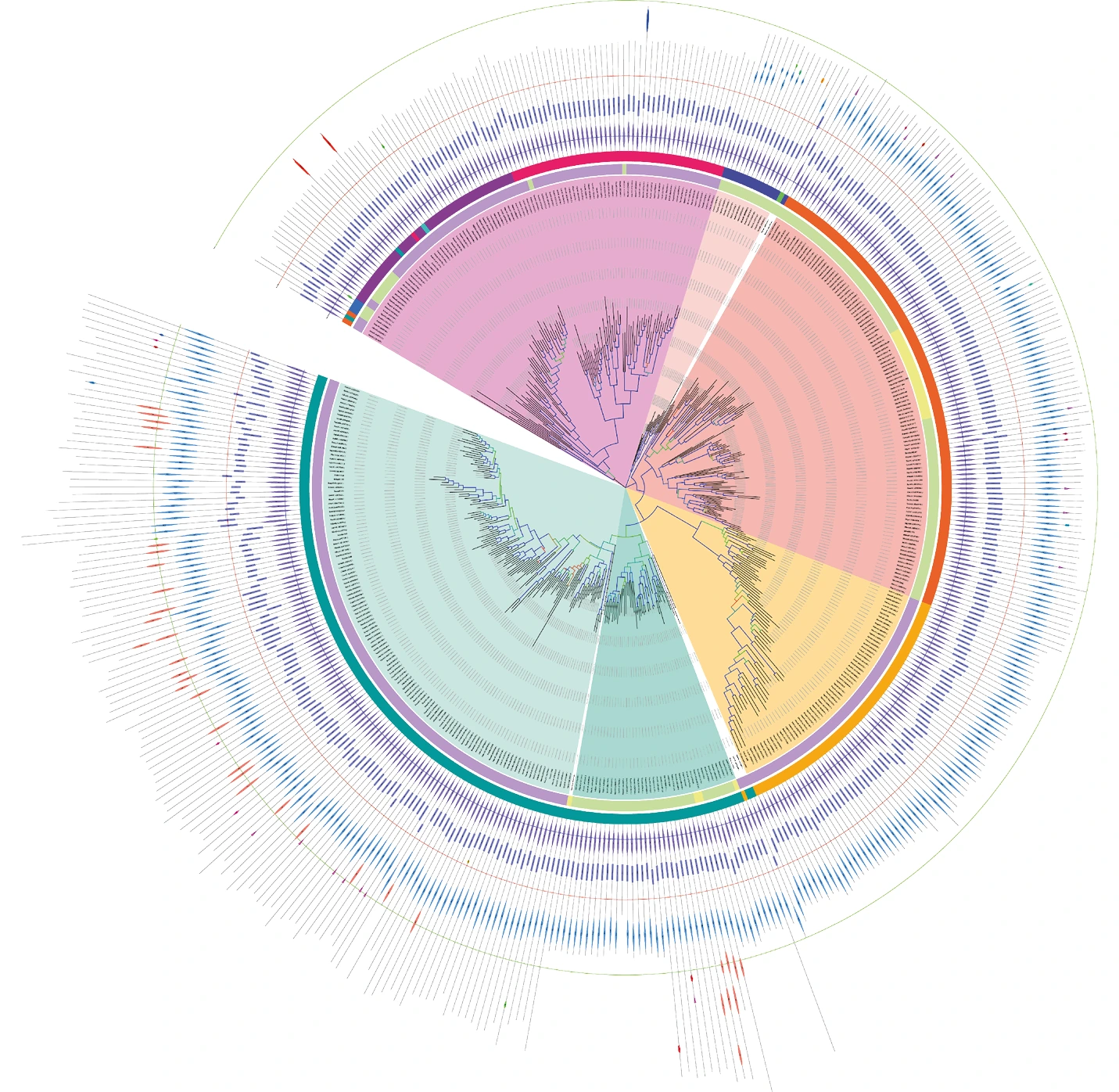
Arbre phylogénétique réalisé à partir de séquences protéiques. Arbre phylogénétique représentant les liens de parenté entre différentes séquences de protéines d’archées et de bactéries. L’arbre est représenté sous forme circulaire, chaque feuille représente une séquence. En passant des feuilles au centre de l’arbre nous remontons le temps. Au centre se trouve la racine, le dernier ancêtre commun. Chaque nœud (croisement) représente le dernier ancêtre commun hypothétique des feuilles regroupées sous ce nœud. La somme des longueurs des branches est proportionnelle au nombre de mutations (différences) observées entre deux séquences, donc au temps évolutif écoulé.
Avec des méthodes bio-informatiques (programmes informatiques de traitement des données biologiques), nous étudions la diversité entre espèces ou entre individus afin de comprendre les mécanismes moléculaires impliqués dans l’adaptation des bactéries à leur environnement.
Etape 1 : comparer les séquences Nous utilisons des séquences nucléotidiques (ADN ou ARN) ou des séquences d’acides aminés (AA – protéines) issues des programmes de séquençage et disponibles pour la communauté scientifique dans des banques de données. Chaque résidu (nucléotide pour l’ADN = A, T, C ou G : acide aminé pour les protéines) est représenté par une « lettre ».
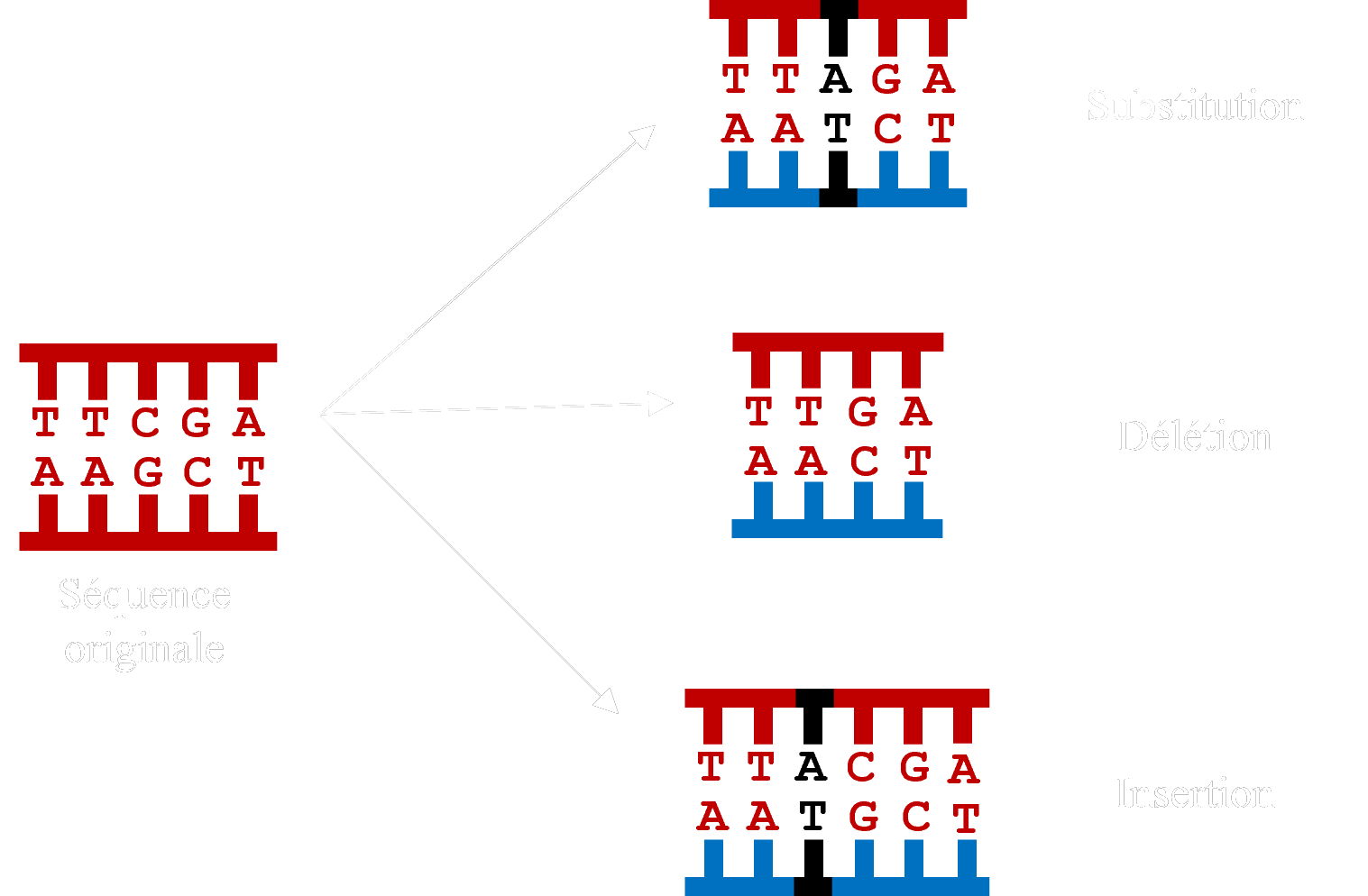
Schéma des différents types de mutation, en utilisant une séquence d’ADN.
Les séquences évoluent par substitution (remplacement), insertion (ajout) et délétion (suppression) de caractères. Pour comparer les séquences, il est donc nécessaire de les éditer pour aligner sur la même colonne les résidus qui partagent la même origine. Cela se fait par l’introduction d’un caractère spécial ‘-’ à l’aide de logiciels spécifiques quand les résidus ne correspondent pas.
Etape 2 : aligner les séquences Le résultat est un alignement multiple. Il peut être colorié en fonction de la nature des résidus (ex. AA chargés + en rouge) ou en fonction de la conservation des résidus dans la colonne (dégradé de bleu). Les résidus conservés au cours de l’évolution jouent un rôle important pour la structure ou la fonction du gène ou de la protéine. D’autres peuvent être modifiés par des mutations. Dans une protéine, un remplacement par un résidu équivalent (même couleur Fig gauche) n’a généralement aucun effet. Dans certains cas, ces modifications sont bénéfiques et témoignent d’une adaptation à un environnement ou des conditions particulières. Certaines bactéries peuvent ainsi vivre dans des milieux extrêmes (très froids ou très chauds, acides ou contaminés). Elles peuvent devenir résistantes aux antibiotiques, ce qui est un problème majeur de santé publique.
Détails d’un alignement multiple de séquences protéiques
Etape 3 : construire un arbre phylogénétique L’alignement multiple, après suppression des colonnes incorrectement alignées (qui contiennent le caractère «- » sur les figures), est utilisé pour construire un arbre phylogénétique. Sur cette représentation schématique d’un arbre phylogénétique, les feuilles sont les séquences étudiées, les nœuds (croisement) sont les séquences ancêtres hypothétiques des feuilles attachées à ce nœud. La racine de l’arbre correspond au dernier ancêtre commun des séquences présentes sur l’arbre. La somme des longueurs des branches menant d’une feuille à l’autre est proportionnelle au nombre de différences observées entre les séquences de ces feuilles.
Un arbre phylogénétique est une représentation graphique de l’histoire évolutive de la famille de séquences considérées.
Exemple d’un arbre phylogénétique
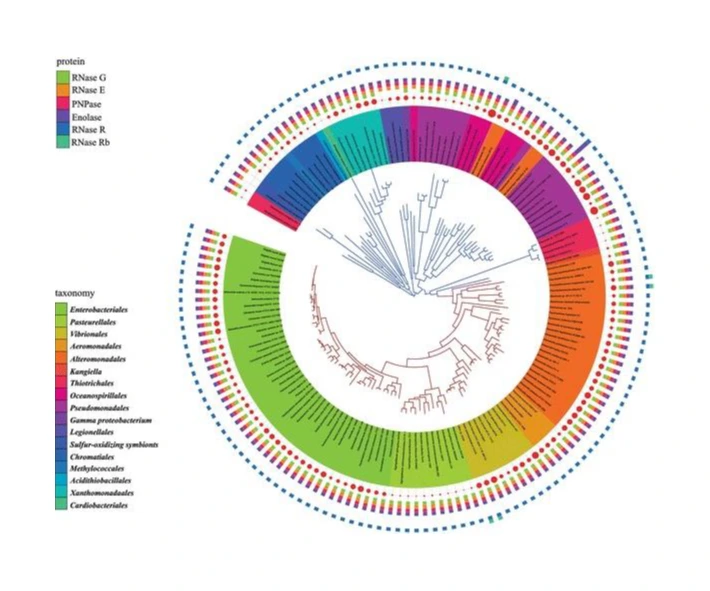
Arbre phylogénétique de la distribution des RNases (Aït Bara et al.,2015 Mol Genet Genomics)
L’arbre obtenu modélise les mutations observées entre les séquences de la famille considérée. Des annotations sont ajoutées de l’intérieur vers l’extérieur du cercle, pour indiquer l’origine taxonomique (méthode de classification des bactéries : nom, famille, etc) et des espèces listées en bas à gauche (couleur des feuilles), la taille des génomes (ronds rouges proportionnels à la taille du génome) et la présence de gènes codant pour une liste de protéines (indiquées en haut à gauche). Il est utilisé pour modéliser les gains et pertes de gènes au cours de l’évolution.
A noter : les mêmes méthodes sont utilisées pour étudier l’évolution de l’espèce humaine, des mammifères, des végétaux, etc.