Avec des méthodes bio-informatiques, nous étudions la diversité génétique entre espèces, afin de comprendre comment les bactéries s’adaptent à leur environnement.
Construction d’un arbre phylogénétique Les données moléculaires que nous utilisons sont des séquences d’ADN ou de protéines pouvant dériver d’une même séquence ancêtre et que nous récupérons dans des banques de données publiques. Avec des analyses bio-informatiques d’alignement de séquences multiples, les résidus (nucléotides ou acides aminés) qui composent ces séquences sont mis en correspondance et mettent en lumière les mutations apparues au cours de l’évolution. En effet, les résidus conservés au cours de l’évolution jouent un rôle important pour la structure ou la fonction du gène ou de la protéine, tandis que d’autres peuvent être modifiés par des mutations. Dans certains cas, ces modifications sont bénéfiques et témoignent d’une adaptation à un environnement ou des conditions particulières. Avec des méthodes de phylogénie moléculaire, un arbre phylogénétique est reconstruit sur la base des différences observées au sein de l’alignement multiple obtenu précédemment. Cet arbre phylogénétique est une représentation graphique qui modélise l’histoire évolutive de la famille de séquences considérée. Ses feuilles correspondent aux séquences considérées ; la longueur de ses branches est proportionnelle à la distance évolutive ; ses nœuds internes représentent des séquences ancêtres hypothétiques (voir schéma dans la version Tout public).
Recherche de l’évolution de systèmes moléculaires dans divers microorganismes Les systèmes biologiques résultent d’une histoire évolutive complexe, car les partenaires et/ou les relations entre eux peuvent avoir été acquis, perdus ou remplacés au cours de l’évolution. Notre principal projet de recherche est d’utiliser les données génomiques pour clarifier l’histoire qui a façonné les processus cellulaires clés (gènes ancestraux, transferts horizontaux de gènes, gains/pertes de gènes) mais aussi pour fournir des informations fonctionnelles pour le travail expérimental. Nous abordons ces questions en réalisant des analyses génomiques comparatives avec des approches phylogénomiques. Depuis plusieurs années, à travers différentes collaborations avec des biologistes expérimentaux, notre équipe s’est focalisée sur l’étude de l’évolution des machineries de dégradation et de traitement de l’ARN chez les bactéries et les archées en réalisant de nombreuses analyses phylogénomiques sur les différents partenaires (ribonucléases, hélicases, …) de ces machineries. Nous vous présentons ci-dessous quelques exemples de nos travaux.
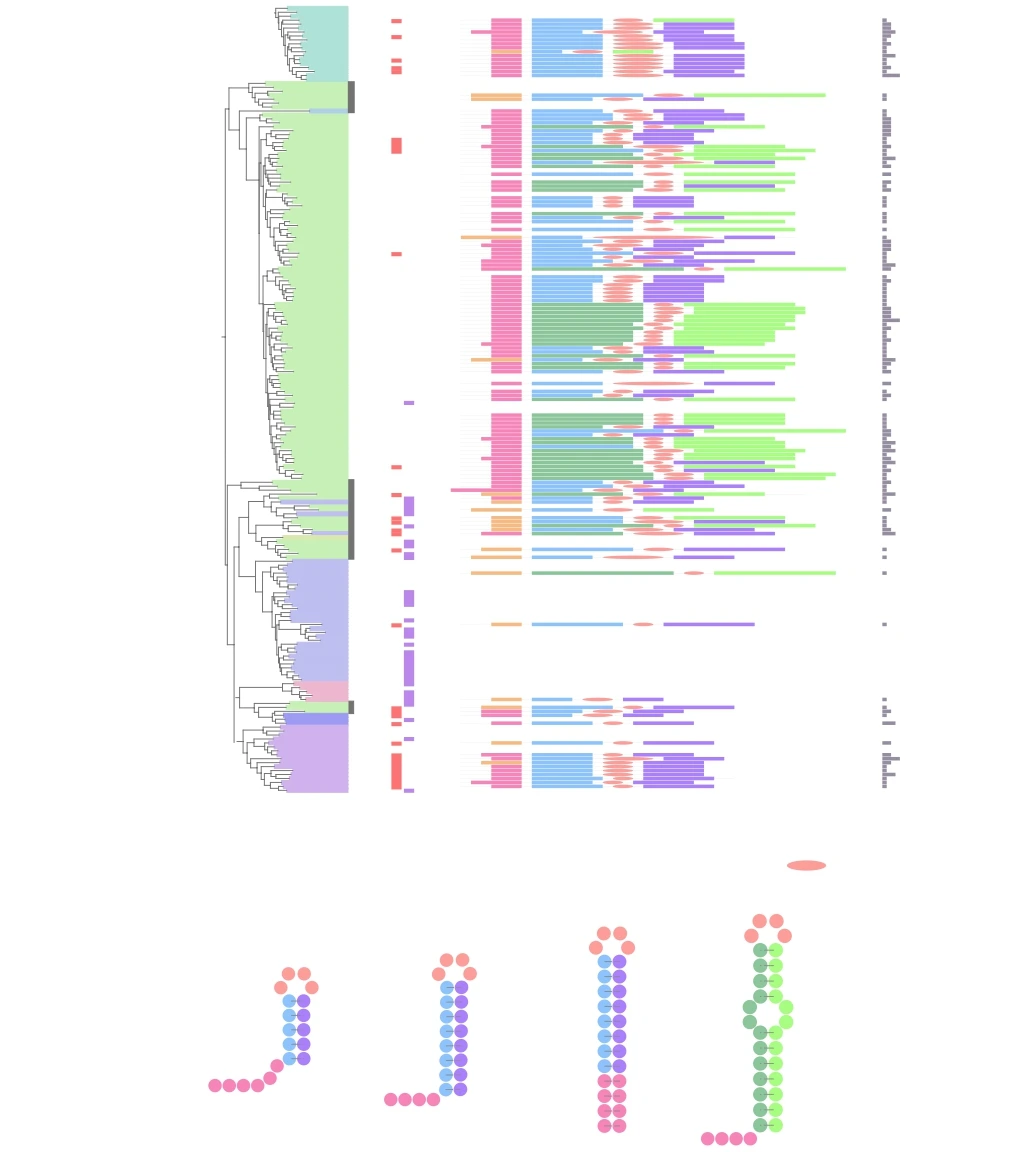
Etude phylogénétique d’une famille d’endonucléases HuH (Quentin et al, 2018, BMC Genomics) Les protéines de la famille TnpAY1 pourraient être responsables de la prolifération des palindromes extragéniques répétés (REP) présents dans de nombreux génomes bactériens formant des structures en épingle à cheveux. Notre objectif était de caractériser la diversité de ces protéines. Nous avons établi une classification détaillée des protéines TnpAY1, consolidée par l’analyse des domaines cœurs conservés et la caractérisation de domaines additionnels (couleurs sur l’arbre sur la partie gauche de la figure). Un domaine fonctionnel est une région de la protéine qui présente une conservation de séquence mise en évidence par alignement multiple auquel nous pouvons associer une fonction. Dans la grande majorité des protéines, nous avons pu identifier des séquences REP (à droite de l’arbre) avec des variations autour de caractéristiques communes confirmant l’implication de ces protéines dans la prolifération des REP dans les génomes. Les données obtenues illustrent la diversité inattendue de la famille TnpAY1 et fournissent un cadre solide pour de futures études évolutives et fonctionnelles. Par leur fonction potentielle dans l’édition de l’ADN simple brin, elles peuvent conférer des réponses adaptatives à la physiologie et au métabolisme de la cellule hôte.
Arbre phylogénétique des séquences de TnpAREP, (Quentin et al, 2018, BMC Genomics)
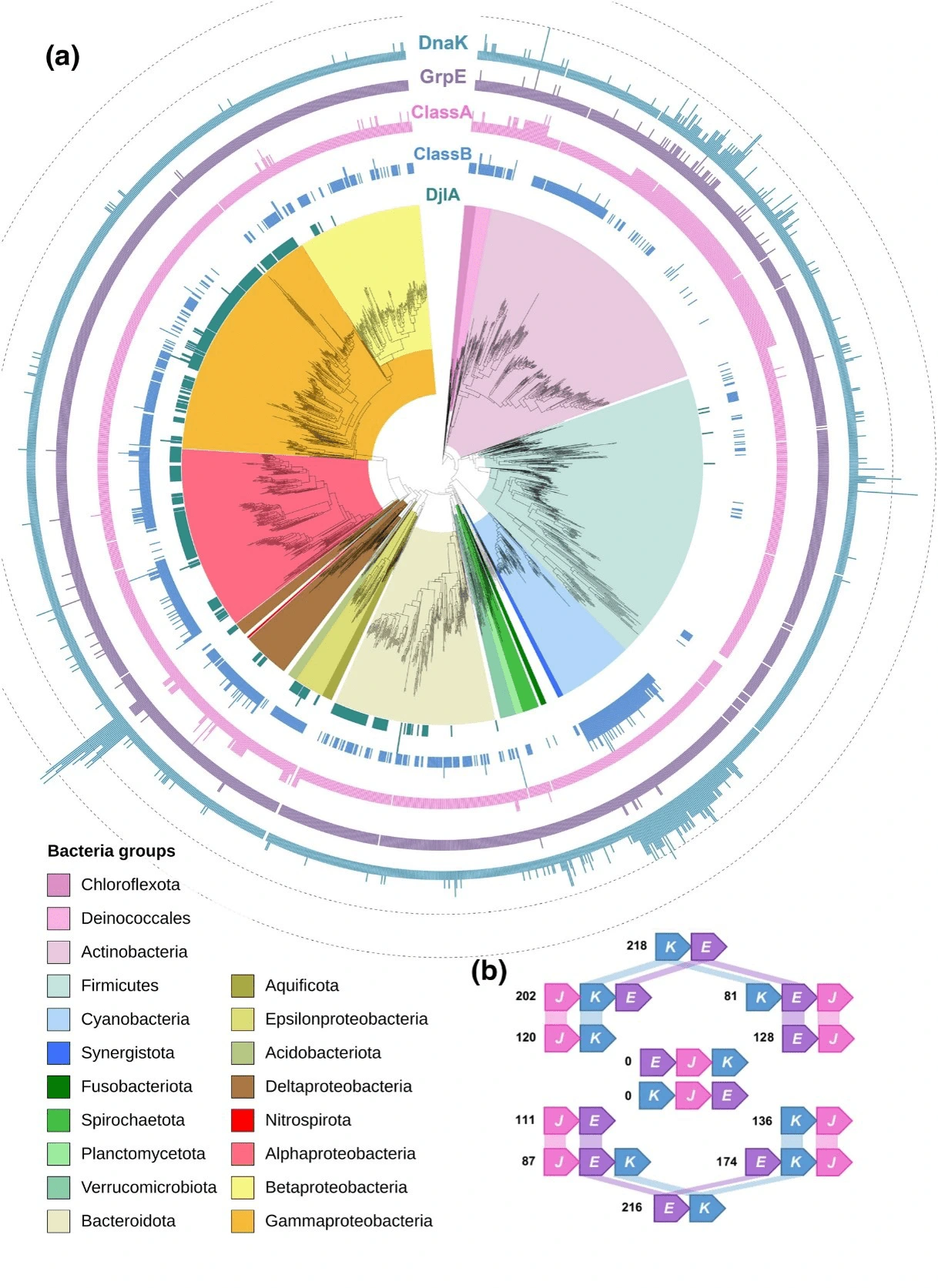
Etude de la distribution taxonomique chez les bactéries des protéines à domaines J (Barriot et al., 2020, J. Mol. Biol.) Les chaperons moléculaires maintiennent l’homéostasie des protéines cellulaires en agissant à presque toutes les étapes des voies de biogenèse des protéines. Le chaperon DnaK/HSP70 a été associé à presque toutes les fonctions chaperonnes essentielles connues chez les bactéries. Pour agir comme un véritable chaperon, DnaK s’appuie strictement sur des partenaires co-chaperons essentiels connus sous le nom de protéines à domaine J (JDP, DnaJ, Hsp40), qui présélectionnent les protéines substrats pour DnaK, lui confèrent sa localisation cellulaire spécifique et stimulent à la fois sa faible activité ATPase et le transfert de substrats. Fait remarquable, le séquençage de génomes a révélé la présence de plusieurs paires de chaperons/co-chaperons JDP/DnaK dans un certain nombre de génomes bactériens, ce qui suggère que certaines paires ont évolué vers des fonctions plus spécifiques. Dans cette étude, nous avons utilisé des ensembles représentatifs de génomes bactériens pour explorer la distribution des chaperons JDP/DnaK chez les bactéries. Cette analyse a révélé un réservoir inattendu de nouvelles protéines à domaine J avec des fonctions très diverses et inexplorées qui seront discutées.
Arbre phylogénétique des séquences de TnpAREP, (Quentin et al, 2018, BMC Genomics)
Références bibliographiques :
– Hajj M, Langendijk-Genevaux P, Batista M, Quentin Y, Laurent S, Capeyrou R, Abdel-Razzak Z, Flament D, Chamieh H, Fichant G, et al. Phylogenetic Diversity of Lhr Proteins and Biochemical Activities of the Thermococcales aLhr2 DNA/RNA Helicase. Biomolecules. 2021; 11(7):950. https://doi.org/10.3390/biom11070950 – Aït-Bara S, Carpousis AJ, Quentin Y. RNase E in the γ-Proteobacteria: conservation of intrinsically disordered noncatalytic region and molecular evolution of microdomains. Mol Genet Genomics. 2015;290:847–62. – Quentin Y, Siguier P, Chandler M, Fichant G. Single-strand DNA processing: phylogenomics and sequence diversity of a superfamily of potential prokaryotic HuH endonucleases. BMC Genomics. 2018 Jun 19;19(1):475. doi: 10.1186/s12864-018-4836-1. PMID: 29914351; PMCID: PMC6006769. – Barriot R, Latour J, Castanié-Cornet MP, Fichant G, Genevaux P. J-Domain Proteins in Bacteria and Their Viruses. J Mol Biol. 2020 Jun 12;432(13):3771-3789. doi: 10.1016/j.jmb.2020.04.014.